Around 5,000 Americans are diagnosed annually with amyotrophic lateral sclerosis (ALS), a disease with a devastatingly short survival period of two to five years post-diagnosis. ALS, a neurodegenerative condition, leads to the progressive death of motor neurons in the brain and spinal cord, manifesting in muscle weakness, breathing difficulties, and sometimes cognitive decline. Despite the severity of ALS, the early triggers that initiate motor neuron breakdown remain largely mysterious, complicating efforts to identify effective treatments.
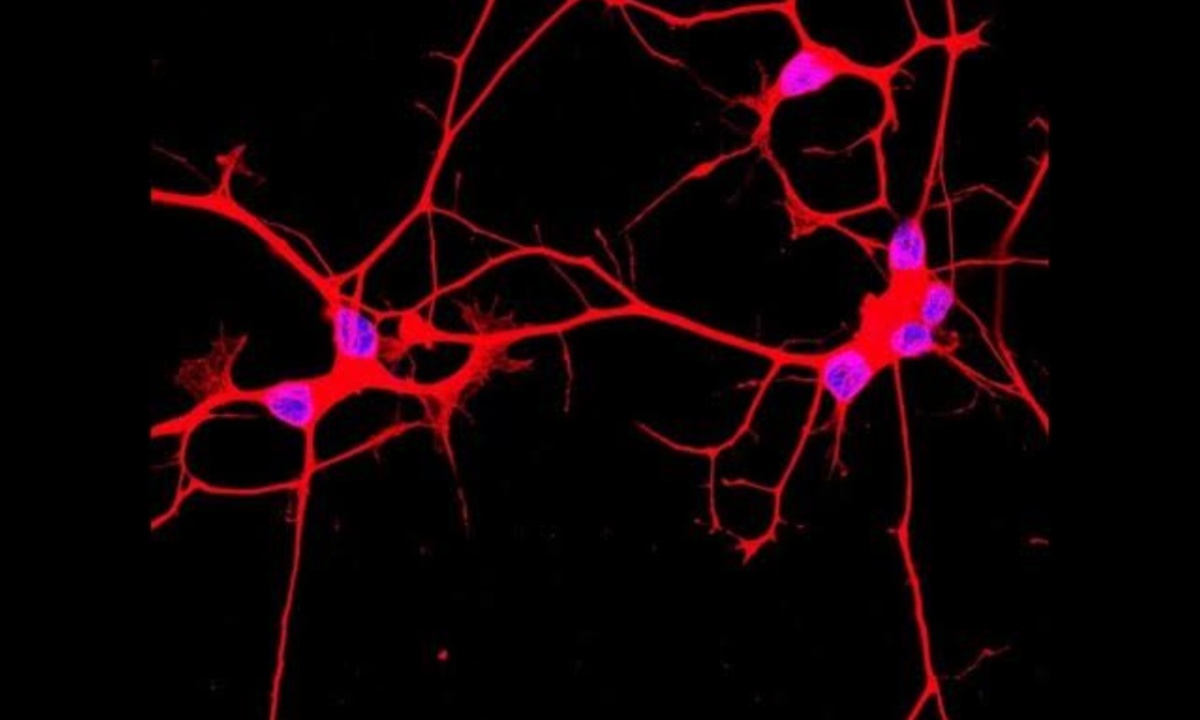
A team of researchers from the University of California San Diego has recently made a breakthrough in understanding what might initiate this neurodegeneration. Published in *Neuron* on October 31, 2024, the study identifies a key pathway that sets off the initial stages of ALS. Their findings revolve around a protein called TDP-43, typically found in the nucleus, which is known to signal ALS when it accumulates in the cytoplasm instead. The UC San Diego team focused on uncovering why TDP-43 ends up in the cytoplasm, a question that has long puzzled scientists.
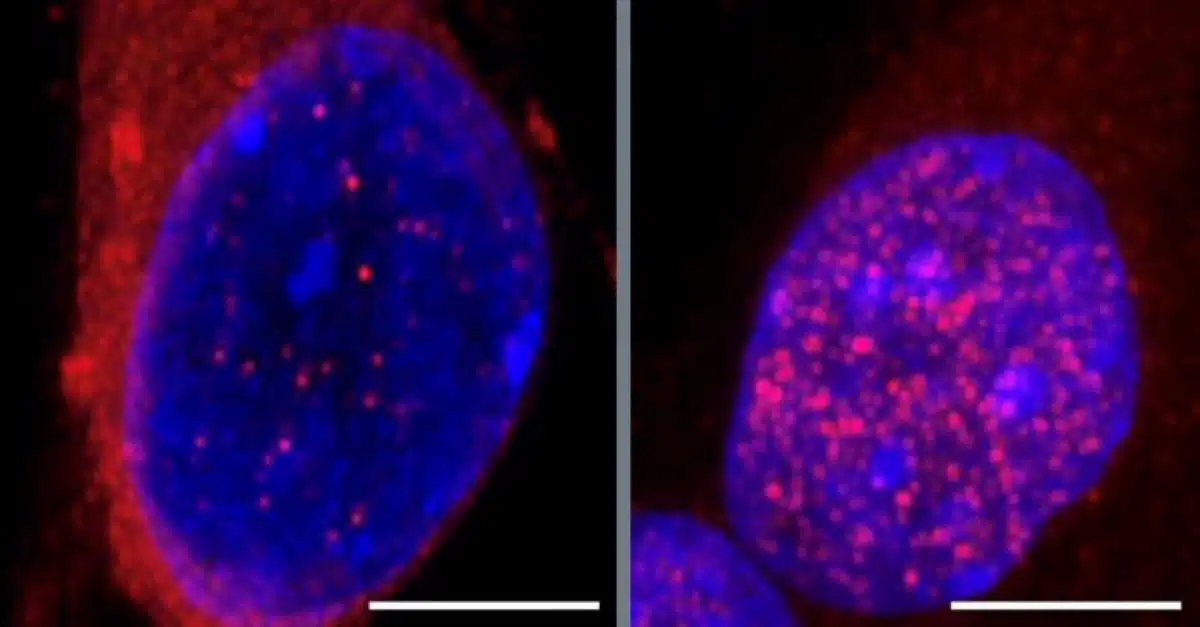
The researchers, led by Dr. Gene Yeo, discovered that the accumulation of a protein called CHMP7 in the nucleus triggers the release of TDP-43 into the cytoplasm, initiating neuron damage. CHMP7 is typically found in the cytoplasm, but in ALS, it builds up in the nucleus, which disrupts cellular processes. By examining which RNA-binding proteins influence this mislocalization, the team identified 55 proteins that may play a role, with 23 of these showing a specific connection to ALS. Targeting several of these proteins increased the CHMP7 buildup, emphasizing their role in ALS progression.
One of the most significant findings emerged from studying the SmD1 protein, which, when reduced, led to the highest levels of CHMP7 buildup in the nucleus. SmD1, an RNA-splicing protein, had not been previously linked to CHMP7 mislocalization. The researchers further observed that this CHMP7 accumulation disrupts nucleoporins, which control the flow of proteins and RNA between the nucleus and cytoplasm. This disruption allows TDP-43 to leave the nucleus, causing it to accumulate in the cytoplasm and disrupting gene regulation essential for neuron health. Increasing SmD1 levels, however, returned CHMP7 to the cytoplasm, restoring cellular balance and preventing neuron damage.
The study opens a potential new treatment avenue by linking the SmD1 protein to ALS pathology. SmD1 is part of the SMN protein complex, known to be defective in spinal muscular atrophy (SMA), another neurodegenerative disease. SMA therapies, such as risdiplam, work by enhancing SMN production, hinting that similar treatments could slow ALS progression. The research team plans to further investigate SMN-targeted therapies for ALS, with the hope that boosting SMN levels in the early stages of ALS could halt the spread of neuron damage and prolong patient survival.